spesim workflow: basic → intermediate → advanced
Source:vignettes/spesim-workflow.Rmd
spesim-workflow.RmdThis vignette gives a structured overview of what spesim can do, arranged as a progression from basic (fast, minimal outputs) through intermediate (common teaching / exploration workflows) to advanced (full reporting and diagnostic panels).
It is meant as a map of the package: which functions are involved, what inputs and outputs to expect, and which “knobs” to turn at each stage.
Big picture workflow
Below is a compact workflow diagram showing the main pipeline.
┌──────────────────────────────┐
│ 1) Configuration │
│ - load_config(init_file) │
│ - or tweak list P in R │
└───────────────┬──────────────┘
│
v
┌──────────────────────────────┐
│ 2) Run simulation │
│ spesim_run() │
│ - creates domain (or use) │
│ - simulates individuals │
│ - places quadrats │
│ - builds matrices │
└───────────────┬──────────────┘
│
v
┌──────────────────────────────────────────────────────┐
│ 3) Primary outputs (returned in-memory; optional disk)│
│ res$species_dist (individual points, with species)│
│ res$quadrats (quadrat polygons) │
│ res$abund_matrix (site × species table) │
│ res$env_gradients (gridded env fields) │
│ res$site_coords (quadrat centroids) │
└───────────────┬──────────────────────────────────────┘
│
├───────────── Basic: maps + matrices
│
├───────────── Intermediate: derived analyses
│ - rank–abundance
│ - occupancy–abundance
│ - SAR
│ - distance–decay
│ - rarefaction
│
└───────────── Advanced: panels + report
- generate_advanced_panel(res)
- generate_full_report(res)Three levels of use
Level 1 — Basic: fast, minimal, robust
Goal: run a small simulation quickly and get the essential objects back for plotting and teaching.
What you typically do
library(spesim)
#> spesim 0.5.2 loaded - try spesim_run() to generate a simulation.
# Start from a known-working example init file
init <- system.file("examples/spesim_init_basic.txt", package = "spesim")
P <- load_config(init)
#> ========== INITIALISING SPATIAL SAMPLING SIMULATION ==========
# Keep it fast (in-memory only)
P$ADVANCED_ANALYSIS <- FALSE
res <- spesim_run(P, write_outputs = FALSE, seed = P$SEED)
#> spesim: running simulation
#> Note: no non-1 interactions for species: A, B, C, D, E, F, G, H, I, J.
#> ---- Interactions Summary ----
#> Species: 10 (A..J)
#> Radius : 0
#> Non-1 entries: 0 (0.0% of 100)
#>
#> Matrix ('.' = 1):
#> A B C D E F G H I J
#> A . . . . . . . . . .
#> B . . . . . . . . . .
#> C . . . . . . . . . .
#> D . . . . . . . . . .
#> E . . . . . . . . . .
#> F . . . . . . . . . .
#> G . . . . . . . . . .
#> H . . . . . . . . . .
#> I . . . . . . . . . .
#> J . . . . . . . . . .
#> ------------------------------
names(res)
#> [1] "P" "domain" "species_dist" "quadrats"
#> [5] "env_gradients" "abund_matrix" "site_env" "site_coords"Useful objects at this level
-
res$domain— sampling domain polygon -
res$species_dist— individuals as sf POINTS with aspeciescolumn -
res$quadrats— quadrat polygons withquadrat_id -
res$abund_matrix— site × species abundance table
Basic visualisations
# Domain + quadrats
plot_quadrats(res$domain, res$quadrats, title = "Quadrats")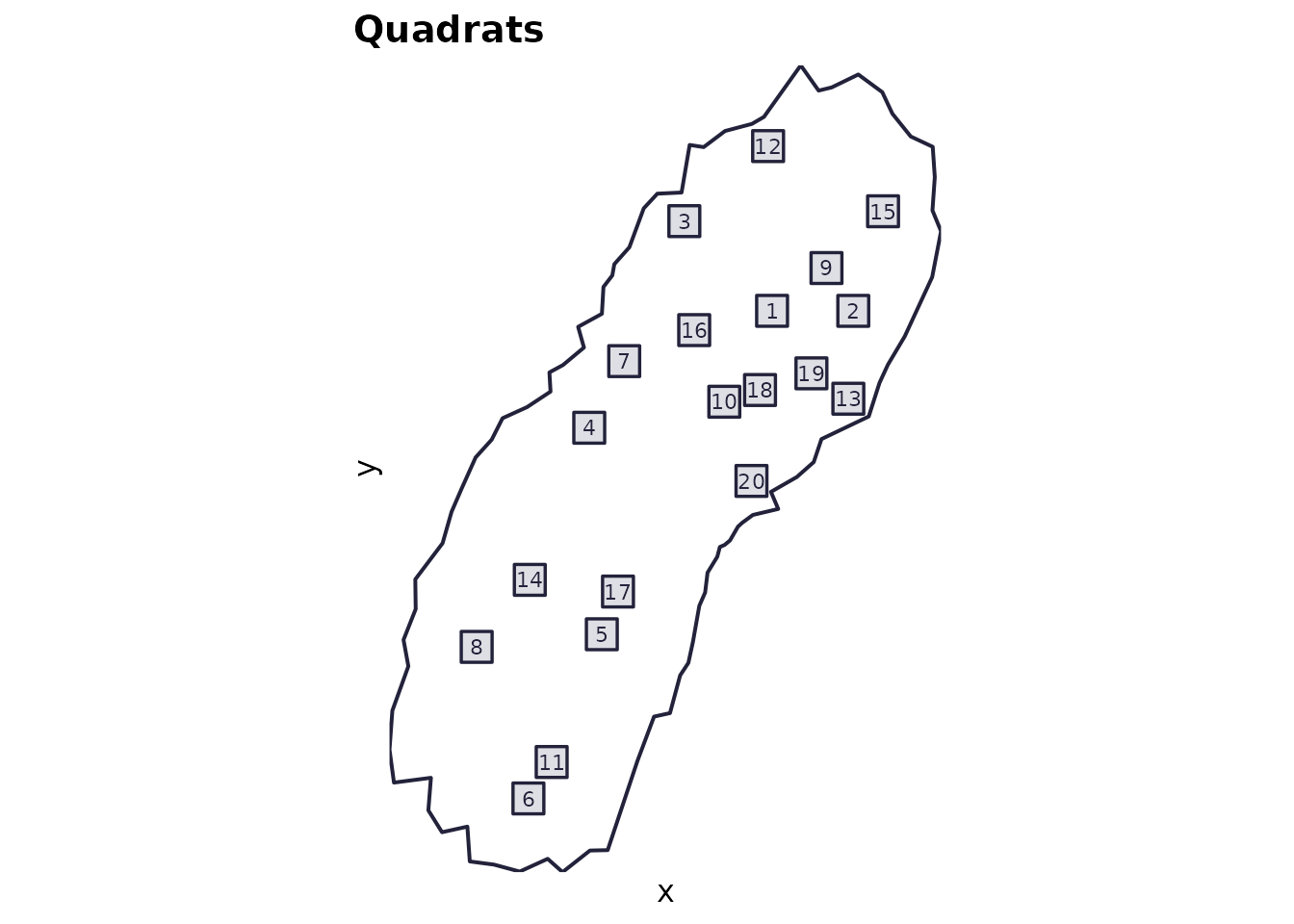
Level 2 — Intermediate: typical analyses + teaching plots
Goal: use the returned objects to compute common community-ecology summaries.
These are the same components that appear in the “advanced panel”, but here you can compute/plot them separately or integrate them into lessons.
Typical intermediate steps:
-
Rank–abundance (SAD):
calculate_rank_abundance(res$species_dist, res$P)plot_rank_abundance(...)
-
Occupancy–abundance:
calculate_occupancy_abundance(res$abund_matrix)plot_occupancy_abundance(...)
-
Species–area curve (SAR):
calculate_species_area(res$abund_matrix)plot_species_area(...)
-
Distance–decay:
calculate_distance_decay(res$abund_matrix, res$site_coords)plot_distance_decay(...)
-
Rarefaction:
calculate_rarefaction(res$abund_matrix)plot_rarefaction(...)
Example (one intermediate product):
ra <- calculate_rank_abundance(res$species_dist, res$P)
plot_rank_abundance(ra)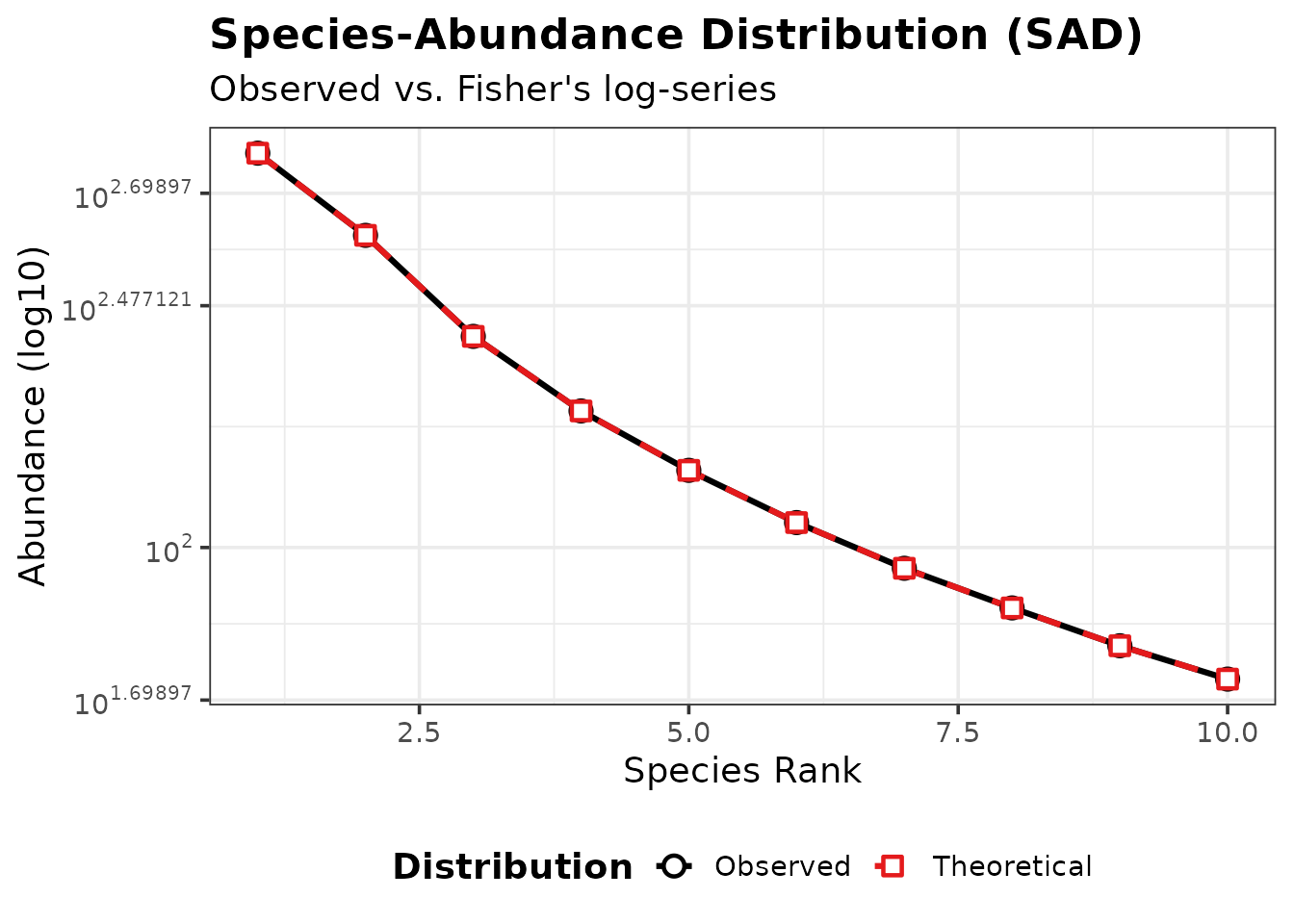
Level 3 — Advanced: full diagnostics + report
Goal: generate a compact multi-plot diagnostic panel and a human-readable report.
There are two ways to do this:
- In-memory (useful for notebooks and teaching):
panel <- generate_advanced_panel(res)
print(panel)
cat(generate_full_report(res))- Write to disk (useful for “run it and inspect outputs” workflows):
# This will write timestamped outputs under out/demo_YYYYMMDD... etc.
res2 <- spesim_run(P, write_outputs = TRUE, output_prefix = "out/demo", seed = P$SEED)What features live where?
Configuration options
- “Init file” workflow:
load_config("path/to/init.txt") - In-memory workflow:
P <- load_config(example); P$N_SPECIES <- 10; ...
Key configuration families:
-
Community size + SAD:
N_SPECIES,N_INDIVIDUALS,DOMINANT_FRACTION,FISHER_ALPHA,FISHER_X -
Environmental filtering:
GRADIENT_*plusSAMPLING_RESOLUTION,ENVIRONMENTAL_NOISE -
Point processes:
SPATIAL_PROCESS_A,SPATIAL_PROCESS_OTHERSand their parameters -
Interactions:
INTERACTION_RADIUS,INTERACTIONS_EDGELISTorINTERACTIONS_FILE -
Sampling design:
SAMPLING_SCHEMEand scheme-specific parameters
Sampling design: quadrat placement
- random:
place_quadrats() - tiled:
place_quadrats_tiled() - systematic:
place_quadrats_systematic() - transect:
place_quadrats_transect() - Voronoi-based:
place_quadrats_voronoi(show_voronoi=TRUE)for teaching/visualisation
See the dedicated vignette: “Quadrat placement schemes in spesim”.
Teaching progression
A suggested progression for a lab/class:
-
Basic run (small, fast) → inspect
res$species_dist,res$quadrats,res$abund_matrix. - Turn on gradients → show how optima/tolerance shapes spatial pattern.
- Compare sampling schemes → show how design changes the observed matrix.
- Turn on interactions → show how neighbourhood effects alter co-occurrence.
- Run the advanced panel → interpret each diagnostic.
- Read the full report → emphasise reproducibility and “what happened?” narratives.
Reproducibility notes
-
load_config()sets a seed (from the init file) for reproducibility. - Some intermediate analyses (e.g., SAR permutations) use randomness
internally; set
set.seed(...)before calling if you want exact repeatability.