spesim recipes: point processes (and the neutral-theory SAD helper)
Source:vignettes/spesim-recipes-point-processes.Rmd
spesim-recipes-point-processes.RmdThis recipe shows how to:
- switch the spatial point-process model for the dominant species (A) and for the remaining species,
- use the neutral-theory SAD helper
(
SAD_MODEL = "zsm"), - use the high-level model family layer
(
MODEL_FAMILY) for neutral and hybrid recruitment.
The key idea is that these are orthogonal knobs:
-
SAD_MODELcontrols how many individuals each species gets (the rank–abundance curve), -
SPATIAL_PROCESS_*controls where individuals are placed in space.
Base configuration
library(spesim)
library(ggplot2)
P0 <- load_config(system.file("examples/spesim_init_complete.txt", package = "spesim"))
# Keep the vignette fast but still visually clear.
P0$N_SPECIES <- 12
P0$N_INDIVIDUALS <- 500
# Make distance–decay curves more interpretable by using a regular quadrat layout.
P0$SAMPLING_SCHEME <- "tiled"
P0$N_QUADRATS <- 16
P0$QUADRAT_SIZE_OPTION <- "medium"
# Turn off environmental filtering to keep this vignette focused on
# point-process and SAD effects (not gradient-driven turnover).
P0$GRADIENT_SPECIES <- character(0)
P0$GRADIENT_ASSIGNMENTS <- character(0)
P0$GRADIENT_OPTIMA <- NULL
P0$GRADIENT_TOLERANCE <- NULL
# Keep interactions off for clarity
P0$INTERACTION_RADIUS <- 0
P0$INTERACTIONS_FILE <- NULL
P0$INTERACTIONS_EDGELIST <- NULL
# A tiny helper to run a case and build its advanced panel.
run_case <- function(P, title) {
res <- spesim_run(P, write_outputs = FALSE, seed = P$SEED, interactions_print = FALSE)
panel <- generate_advanced_panel(res) + patchwork::plot_annotation(title = title)
list(res = res, panel = panel)
}
rank_df <- function(res, label) {
ra <- calculate_rank_abundance(res$species_dist, res$P)
ra <- subset(ra, Source == "Observed")
ra$Scenario <- label
ra
}
decay_df <- function(res, label) {
dd <- calculate_distance_decay(res$abund_matrix, res$site_coords)
dd$Scenario <- label
dd
}Neutral theory in spesim (current implementation)
spesim supports two neutral-style routes:
-
SAD_MODEL = "zsm": theta-only neutral-like SAD helper. -
MODEL_FAMILY = "neutral_hubbell_like": sequential recruitment with immigration/speciation and dispersal kernel. -
MODEL_FAMILY = "hybrid": same recruitment core plus environmental sorting.
What it can do
-
SAD_MODEL = "zsm"gives neutral-like rank–abundance control viaZSM_THETA. -
MODEL_FAMILY = "neutral_hubbell_like"gives distance–decay that emerges from dispersal-limited recruitment. -
MODEL_FAMILY = "hybrid"blends dispersal limitation with environmental sorting.
What it cannot do (yet)
- It is still a synthetic simulator (not calibrated population dynamics through time).
- Species labels remain fixed (
A..) rather than open-ended speciation lineages. - Recruitment is intentionally lightweight for method testing speed.
The practical takeaway: use SAD_MODEL = "zsm" when you
only want neutral-like abundance structure; use
MODEL_FAMILY = "neutral_hubbell_like" or
"hybrid" when you want spatial patterns to emerge from
recruitment.
Example A: SAD differences (Fisher vs neutral-theory ZSM)
Here we hold the spatial process fixed at Poisson
(CSR) so differences in the SAD plots are driven by
SAD_MODEL.
P_sad <- P0
P_sad$SPATIAL_PROCESS_A <- "poisson"
P_sad$SPATIAL_PROCESS_OTHERS <- "poisson"
# Fisher baseline
P_fisher <- P_sad
P_fisher$SAD_MODEL <- "fisher"
P_fisher$DOMINANT_FRACTION <- 0.30
# Neutral-like SADs via zsm
P_zlow <- P_sad
P_zlow$SAD_MODEL <- "zsm"
P_zlow$ZSM_THETA <- 1.5
P_zlow$ZSM_M <- NA_real_
P_zhigh <- P_zlow
P_zhigh$ZSM_THETA <- 50
case_fisher <- run_case(P_fisher, "Poisson + Fisher SAD")
case_zlow <- run_case(P_zlow, "Poisson + ZSM (theta = 1.5)")
case_zhigh <- run_case(P_zhigh, "Poisson + ZSM (theta = 50)")Rank–abundance (overlay)
ra_all <- rbind(
rank_df(case_fisher$res, "Fisher"),
rank_df(case_zlow$res, "ZSM θ=1.5"),
rank_df(case_zhigh$res, "ZSM θ=50")
)
ggplot(ra_all, aes(Rank, Abundance, colour = Scenario)) +
geom_line(linewidth = 1) +
geom_point(size = 2) +
scale_y_log10() +
labs(
title = "Rank–abundance curves: Fisher vs neutral-theory helper",
y = "Abundance (log10 scale)",
x = "Rank (1 = most abundant)"
) +
theme_minimal(base_size = 12)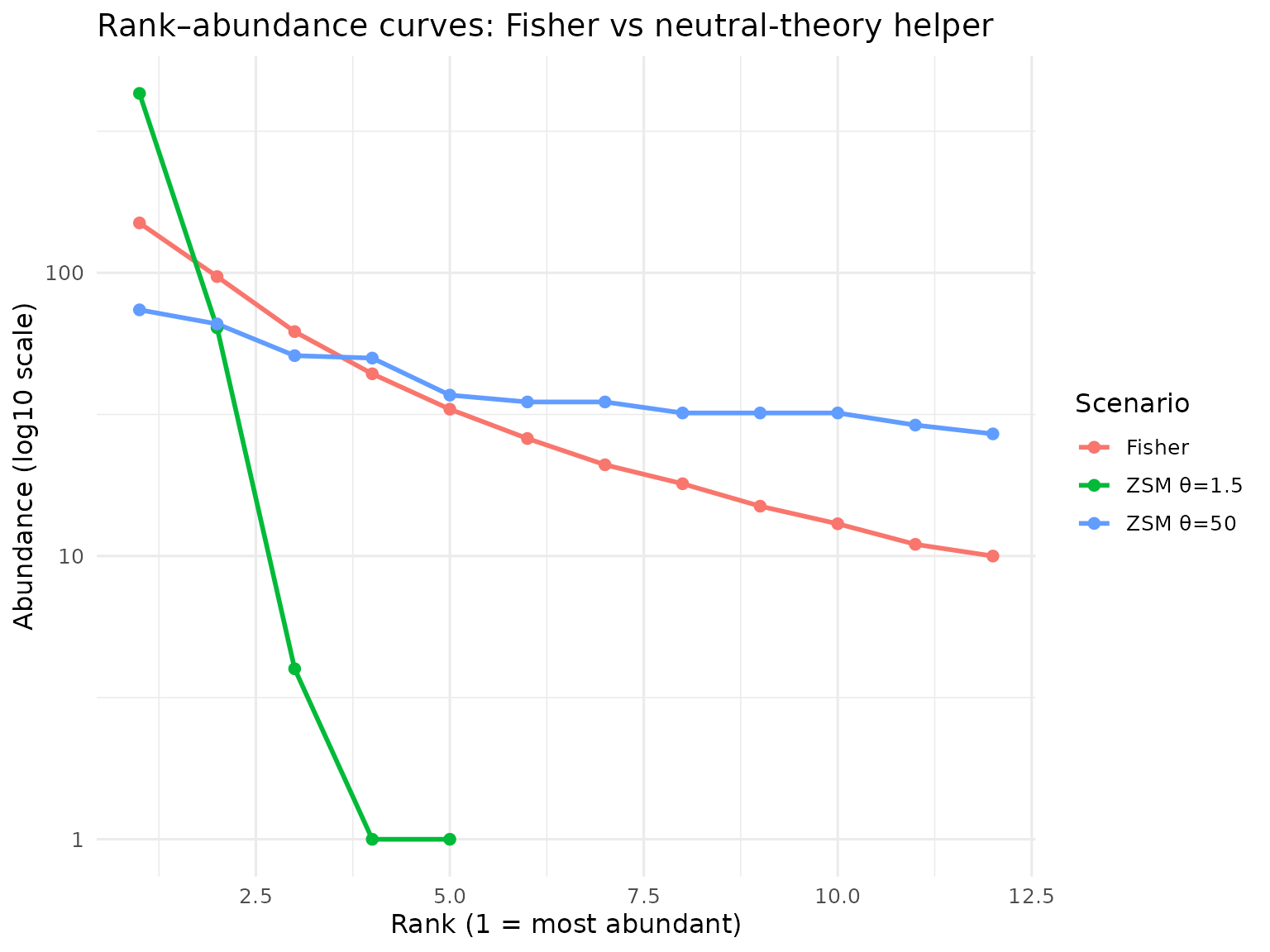
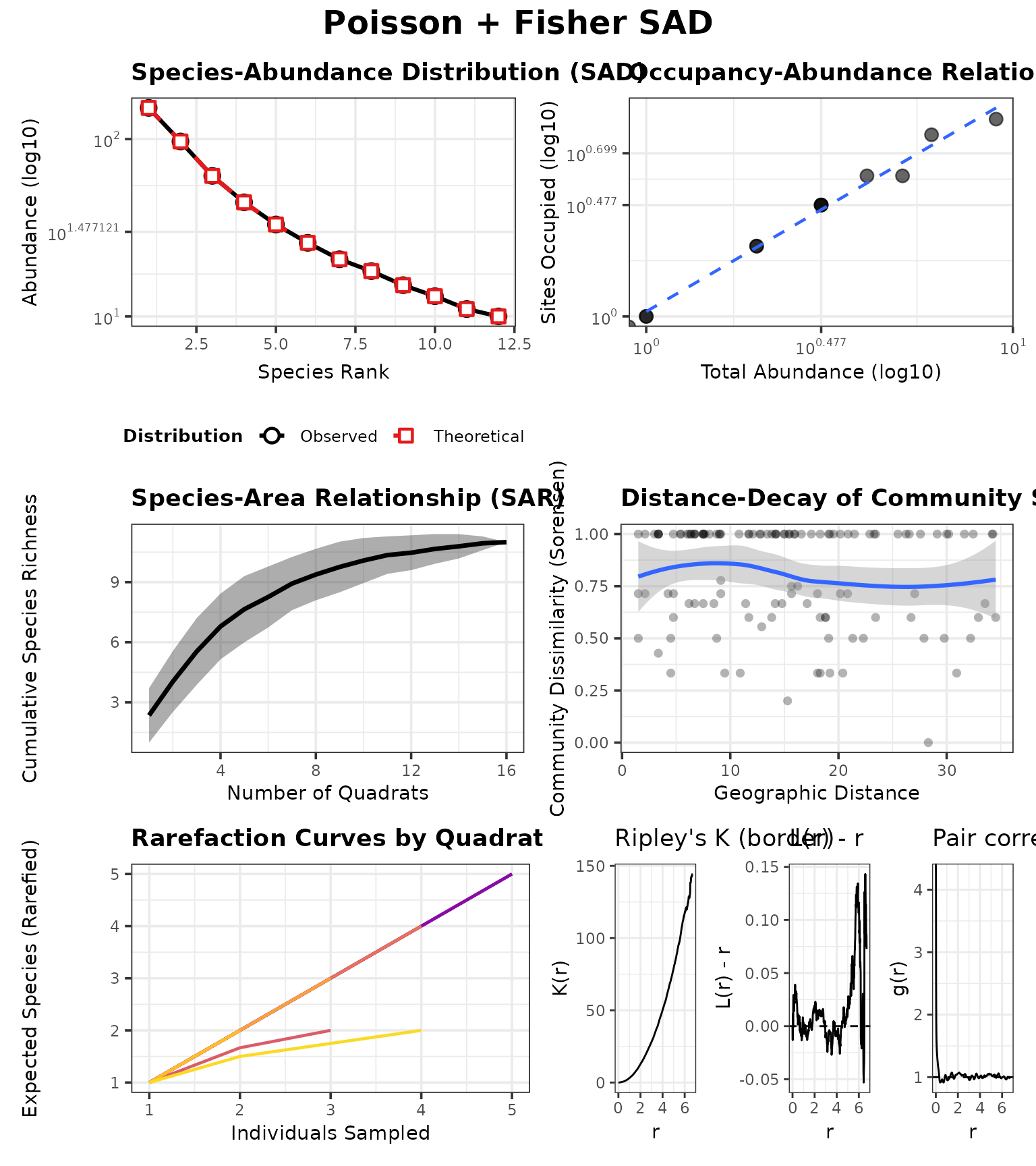
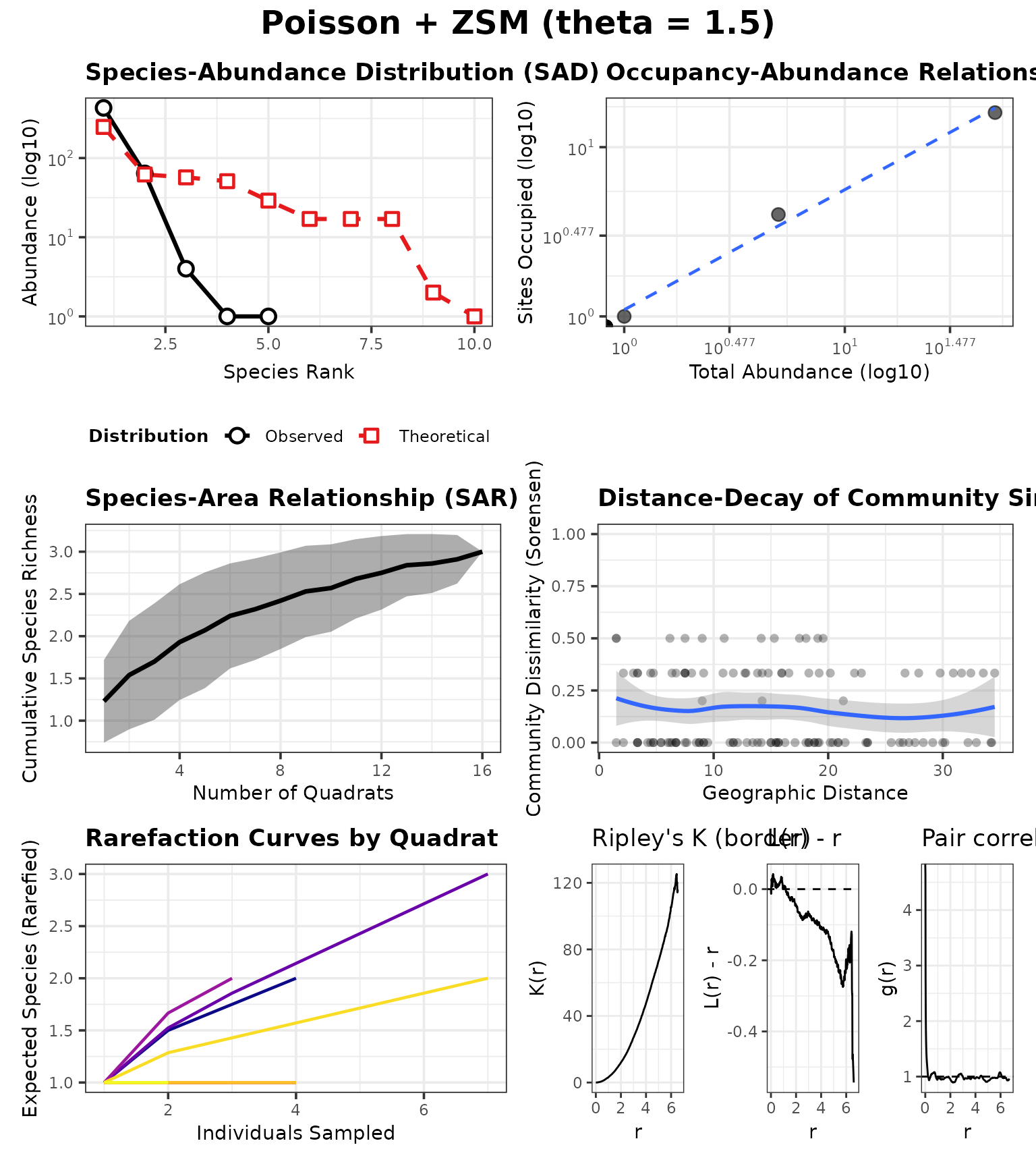
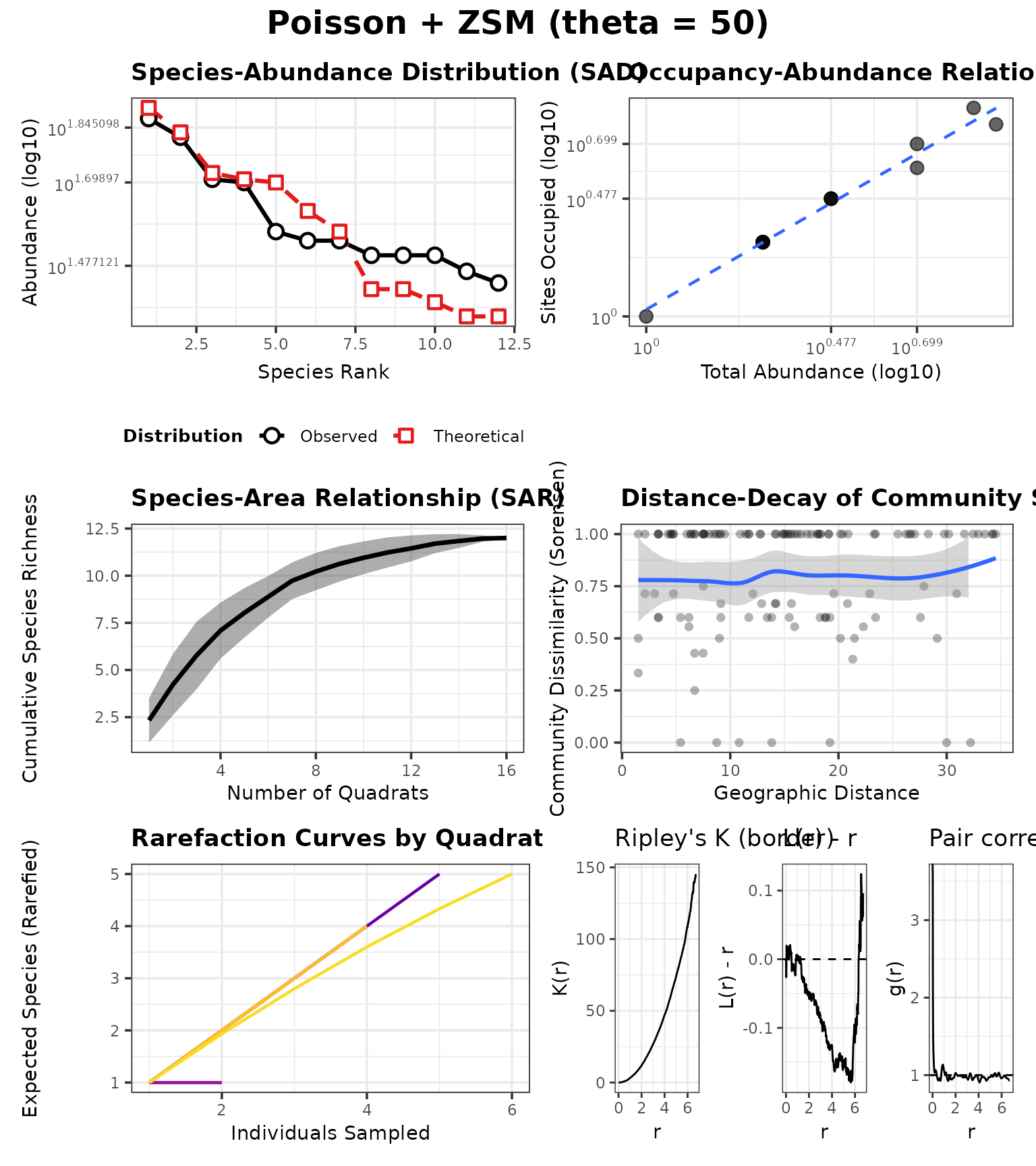
Example B: Spatial differences (distance–decay) under a fixed neutral SAD
Now we fix the SAD model to neutral-theory ZSM and vary only the point-process.
To make the contrast obvious, we choose a strongly structured placement:
- Dominant species A: tight Thomas clustering (many offspring, small sigma)
- Other species: strong Strauss inhibition (repulsion)
P_neutral <- P0
P_neutral$SAD_MODEL <- "zsm"
P_neutral$ZSM_THETA <- 10
P_neutral$ZSM_M <- NA_real_
# CSR baseline
P_neutral_pois <- P_neutral
P_neutral_pois$SPATIAL_PROCESS_A <- "poisson"
P_neutral_pois$SPATIAL_PROCESS_OTHERS <- "poisson"
# Strong spatial structure
P_neutral_struct <- P_neutral
P_neutral_struct$SPATIAL_PROCESS_A <- "thomas"
P_neutral_struct$A_PARENT_INTENSITY <- NA
P_neutral_struct$A_MEAN_OFFSPRING <- 25
P_neutral_struct$A_CLUSTER_SCALE <- 0.45
P_neutral_struct$SPATIAL_PROCESS_OTHERS <- "strauss"
P_neutral_struct$OTHERS_R <- 1.2
P_neutral_struct$OTHERS_STRAUSS_GAMMA <- 0.15
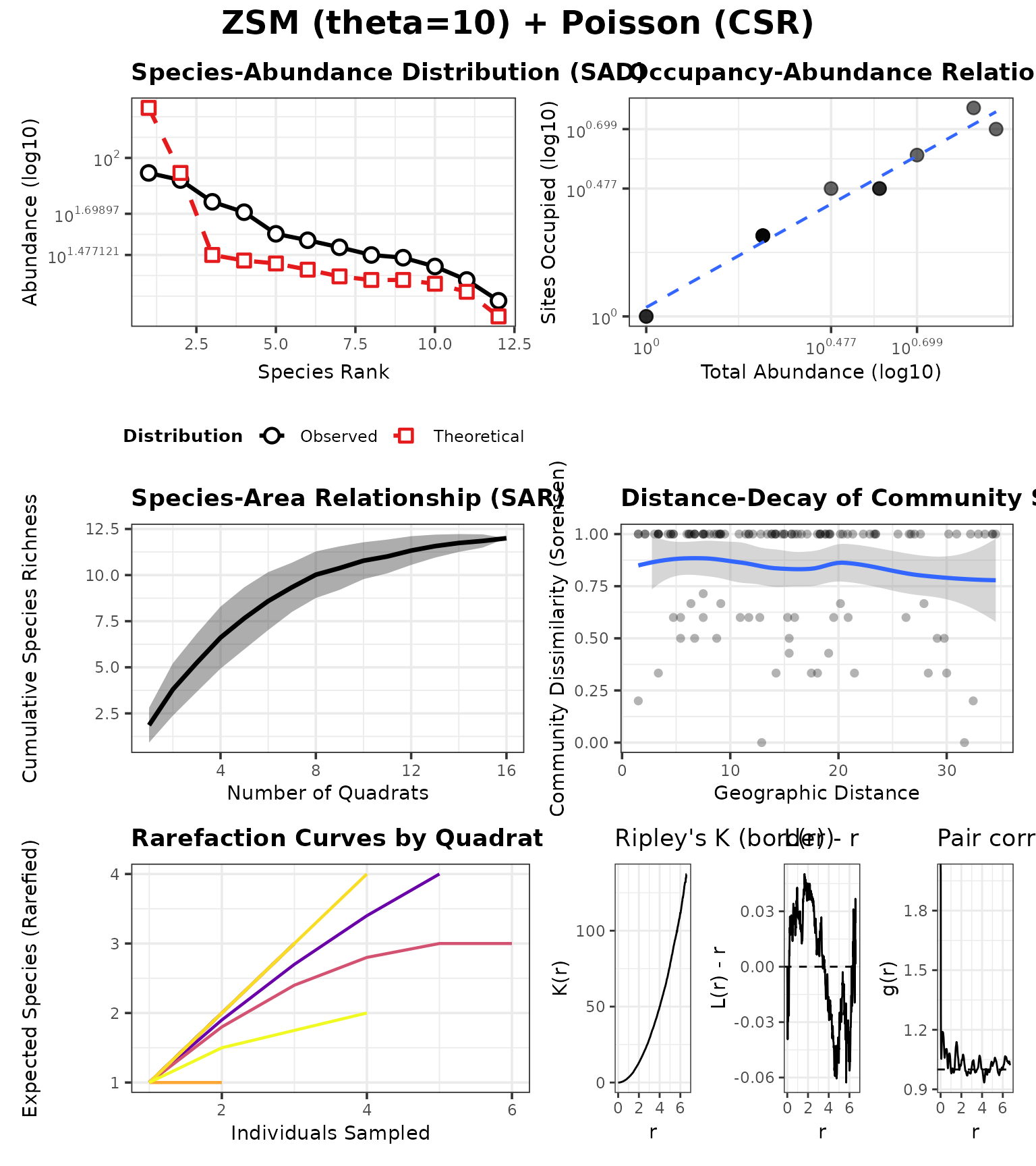
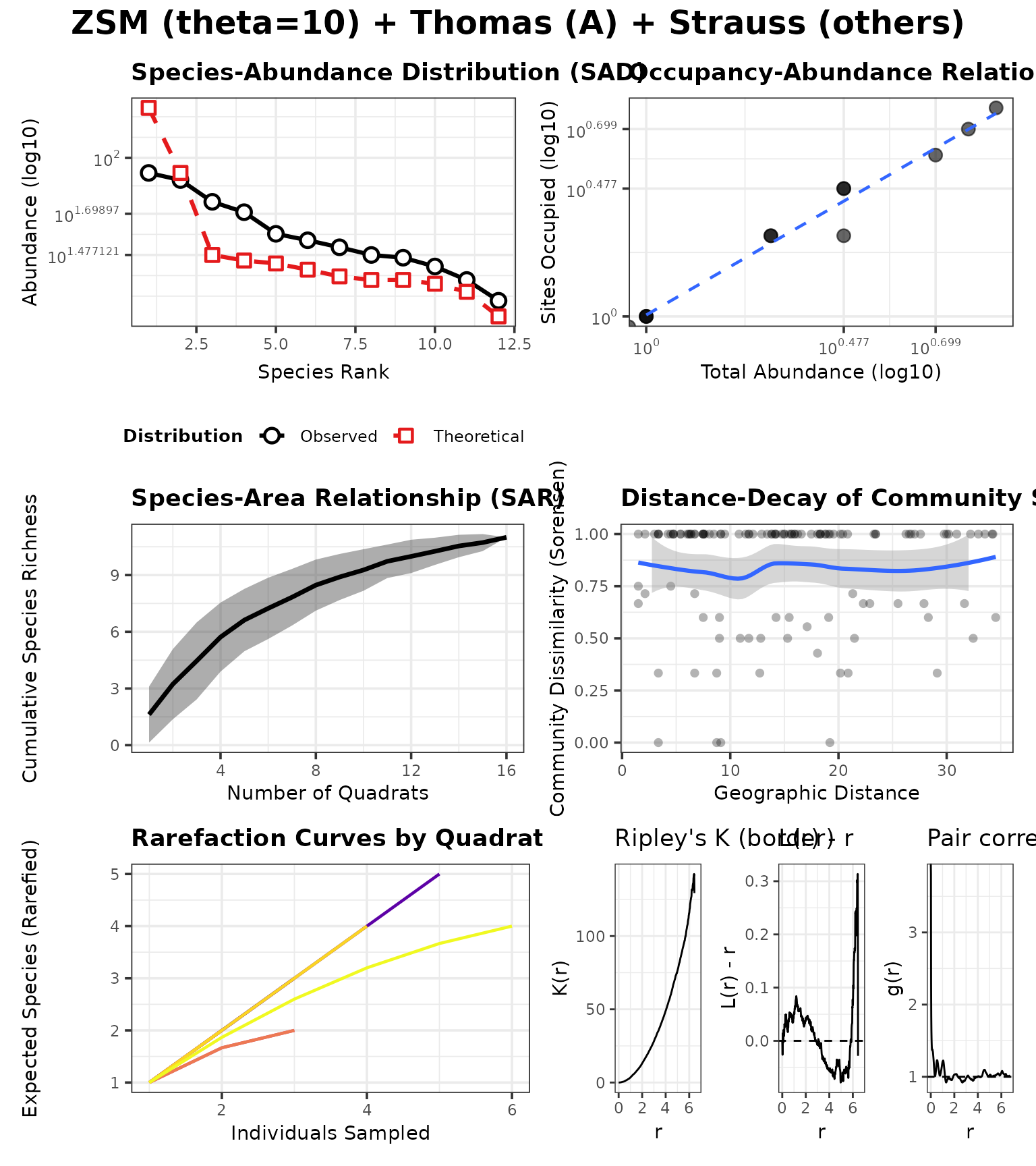
case_neutral_pois <- run_case(P_neutral_pois, "ZSM (theta=10) + Poisson (CSR)")
case_neutral_struct <- run_case(P_neutral_struct, "ZSM (theta=10) + Thomas (A) + Strauss (others)")Distance–decay (overlay)
dd_all <- rbind(
decay_df(case_neutral_pois$res, "Poisson"),
decay_df(case_neutral_struct$res, "Thomas + Strauss")
)
ggplot(dd_all, aes(Distance, Dissimilarity, colour = Scenario)) +
geom_point(alpha = 0.25) +
geom_smooth(se = FALSE, linewidth = 1.2, method = "loess", span = 0.75) +
labs(
title = "Distance–decay under the same neutral SAD",
subtitle = "Spatial structure changes turnover with distance; SAD model is held constant",
x = "Inter-quadrat distance",
y = "Community dissimilarity (Bray–Curtis on presence/absence)"
) +
theme_minimal(base_size = 12)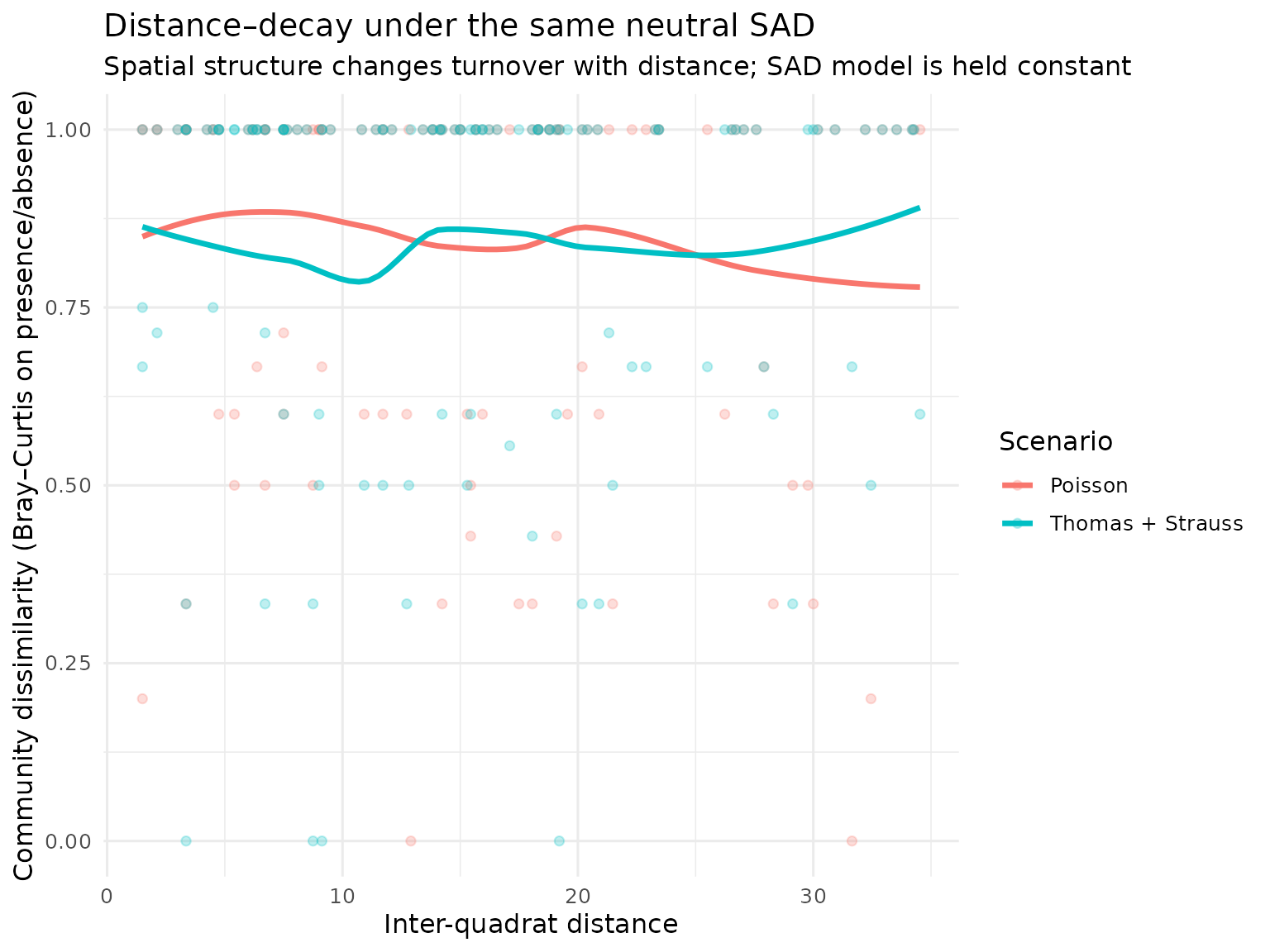
Example C: Model families (neutral_hubbell_like vs hybrid)
Here we switch from point-process assembly to the high-level family layer.
P_family <- P0
P_family$N_INDIVIDUALS <- 400
P_family$SAD_MODEL <- "zsm"
P_family$ZSM_THETA <- 12
P_family$GRADIENT_SPECIES <- c("A", "B", "C", "D")
P_family$GRADIENT_ASSIGNMENTS <- c("temperature", "temperature", "elevation", "rainfall")
P_family$GRADIENT_OPTIMA <- c(A = 0.25, B = 0.70, C = 0.55, D = 0.60)
P_family$GRADIENT_TOLERANCE <- c(A = 0.12, B = 0.12, C = 0.16, D = 0.14)
P_hubbell <- P_family
P_hubbell$MODEL_FAMILY <- "neutral_hubbell_like"
P_hubbell$NEUTRAL_M <- 0.08
P_hubbell$DISPERSAL_KERNEL <- "gaussian"
P_hubbell$DISPERSAL_SCALE <- 0.55
P_hybrid <- P_hubbell
P_hybrid$MODEL_FAMILY <- "hybrid"
P_hybrid$HYBRID_ENV_WEIGHT <- 1.3
case_hubbell <- run_case(P_hubbell, "MODEL_FAMILY = neutral_hubbell_like")
case_hybrid <- run_case(P_hybrid, "MODEL_FAMILY = hybrid")
dd_family <- rbind(
decay_df(case_hubbell$res, "neutral_hubbell_like"),
decay_df(case_hybrid$res, "hybrid")
)
ggplot(dd_family, aes(Distance, Dissimilarity, colour = Scenario)) +
geom_point(alpha = 0.25) +
geom_smooth(se = FALSE, linewidth = 1.2, method = "loess", span = 0.75) +
labs(
title = "Model-family comparison",
subtitle = "Hybrid combines neutral recruitment with environmental sorting",
x = "Inter-quadrat distance",
y = "Community dissimilarity (Bray–Curtis on presence/absence)"
) +
theme_minimal(base_size = 12)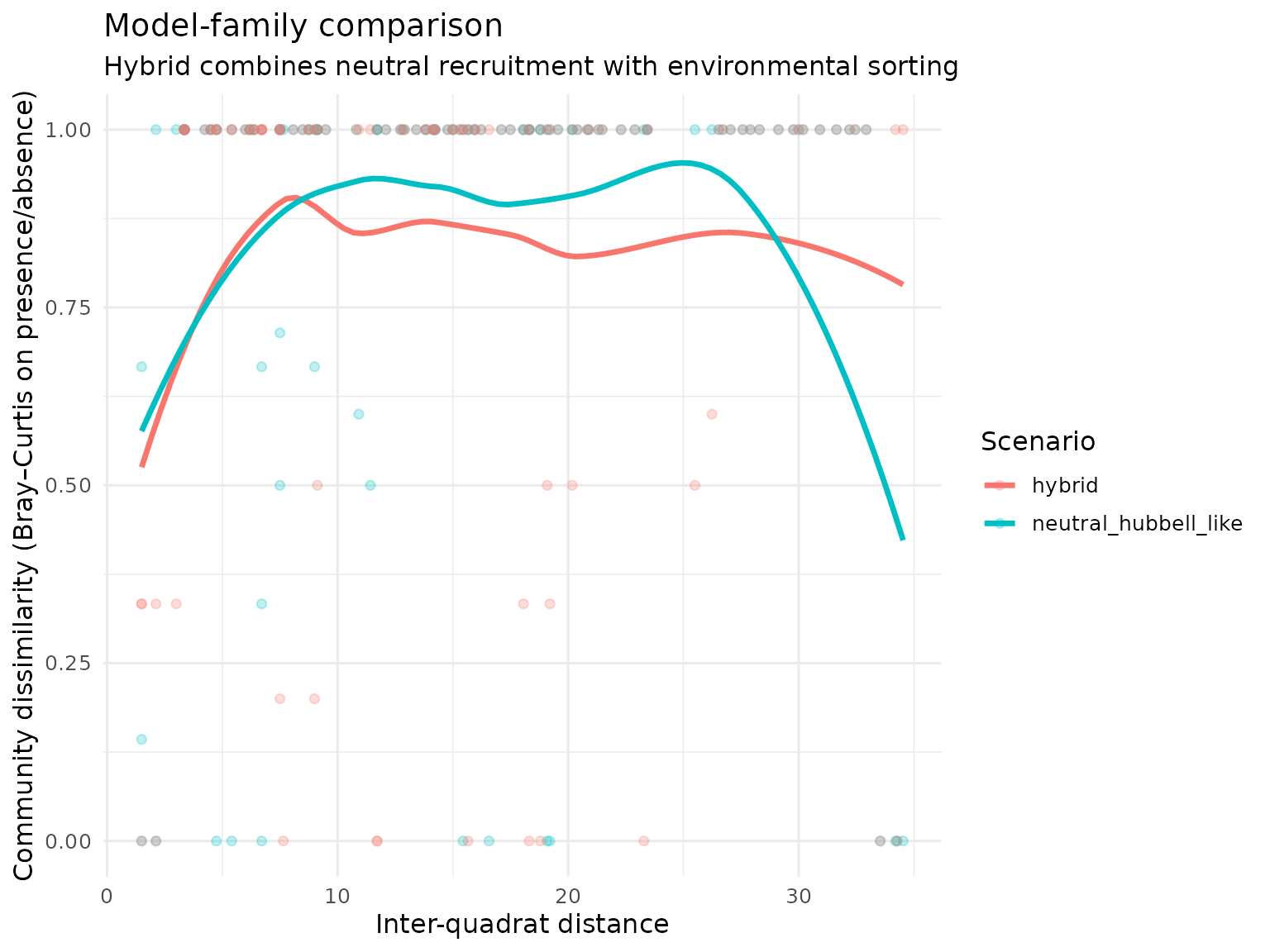
Notes / tips
- Use
MODEL_FAMILY = "manual"when you want full low-level control. - Use
MODEL_FAMILY = "neutral_hubbell_like"or"hybrid"when you want dispersal-driven structure to emerge from recruitment. - For Doubs-style/seaweed-style constrained gradients, set
DOMAIN_TYPE = "network"or"coastline"and useDISTANCE_METRIC = "along_path". - You can replace synthetic raster fields by supplying
ENV_COVARIATES_FILE(section/node-level covariates withx,yplus environmental columns). - If the distance–decay looks noisy, increase
N_QUADRATSand/or reduce quadrat size. - For pedagogy, it helps to vary one knob at a time (SAD vs point process), then show how they interact.